Systems and procedure packs
In other articles I discussed grouping devices into the Basic UDI-DI, and also the strategic importance of the Basic UDI-DI from a regulatory perspective. This article sets out how UDI is used for the identification of systems and procedure packs.
Article 22 defines a system of procedure pack as a combination of at least one CE marked medical device together with other CE-marked medical devices or IVD�s and/or other products that are in conformity with the applicable legislation, where a (legal) person assigns a specific use for that combination and these products are intended to be used together for this particular application.
Obviously, if a device is intended to be used in combination with other devices because it�s interoperable or compatible with them, that is a type of use that is in line with its intended purpose and this does not make it into a system or procedure pack component. But at the moment its use is defined more specifically than the intended purpose of the original device or product in terms of an intended use for the combination, this should be considered a system or procedure pack component.
To understand how this works in practice, let�s look at a simple example, a single use line with a needle with some other products added as the example expands:
1. If the line is intended to be used with a range of needles and the needle can be used with several other devices, including lines, they are each individual compatible medical devices that are intended to be used in combination. They are not part of a system or procedure pack.
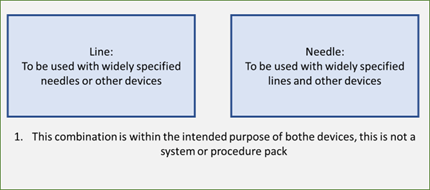
2. If the line and the needle are marketed under their own label and CE-mark but an economic operator puts them in a box together and specifies that this particular line and that particular needle is to be used for a specific procedure, the combination of the two could be seen as a procedure pack.

3. They may also be placed on the market as a single product, with a single label and CE-mark covering both components. The technical documentation, like the clinical evaluation report, must provide evidence for the compliance of this particular combination of components for that particular intended purpose.

A clip can be added to fix the line. That clip is just handy and therefore it is not necessary for the line to be used as intended. Therefore, the clip is not a medical device and it is not an accessory; it does not fall in the scope of the MDR.
4. If the clip is put in the same box as the line. Line plus clip or line, clip and needle can be seen as a procedure pack. This has to be labelled as such.

5. The clip may also be considered part of the line and should then be covered under the label and technical documentation of the line.

The line and needle must be used together with a reusable pump. The pump is controlled by software. This combination can be described in two versions:
6. From the perspective of the line or line procedure pack, the line is described at the level of general specifications and the pump is specified in terms of line diameter and range of volume. These are devices or procedure packs that can be used together, without being labelled as a system.
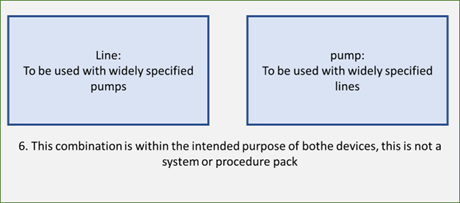
7. In the case this combination is specified up to the level of specific components and marketed together, this is brought within the scope of Article 22. As the single use components and the pump cannot be distributed together, they cannot be seen as a procedure pack; they should be considered a system. The single use components can be placed on the market in any of the above mentioned configurations. There must be a systems statement declaring they can be used together with the specific pump. Note that this may require two levels of statements, one for a procedure pack and one for the system combining the procedure pack with the pump.

Software controlling a device
The pump is hardware that uses software in order to operate and has a label plus UDI. The software on the device may either be used for just controlling it, or it may do calculations that have a significant impact on clinical outcomes (e.g. frequent recalculation of the administrated volume). It is important to fully understand this difference, because updates in the software may have an impact on the UDI.
In the case of a simple fix, this may be reflected in the version number of the software that may be displayed on the display. Strictly speaking this should impact the UDI on the label of the pump, because a patch in software should be reflected in a different UDI-PI. In practice I don�t expect this to be an issue, but strictly speaking, this could result in a non-conformity. But just like Tesla cars one day became self-driving vehicles overnight, thanks to an over-the-air software update, something similar could (and often does) happen to software controlling a medical device. This could go as far as changing the intended purpose and essential design features of the pump to such extend that a new Basic UDI-DI would be necessary. But the UDI on the label still links to the �old� version, and, if not all devices have this new software, it is possible there is confusion about identifying these devices in the distribution chain or in use.
The practical approach for this would be to assign Basic UDI-DI�s to the hardware as well as the software. If the intended purpose of the software is to control that specific piece of hardware and the other way round, both devices are intended to be used together and they are not a system. At the moment the software is updated, this will show in their user interface and there the new UDI-DI and UDI-PI can be displayed, while the UDI on the hardware does not have to change.
In the case the software changes to such degree that a new Basic UDI-DI must be assigned, the intended purpose of the hardware does not cover that version of the software. Then the manufacturer can combine these devices into a system and assign a system UDI to it. In the case of fast developing technology, where it is very likely the software will have significant updates in the foreseeable future, a manufacturer may decide to assign a system UDI to this combination from the start.
Article 22 and UDI
According to Article 29 a system or procedure pack must have their own Basic UDI-DI assigned, and the core data elements must be entered into EUDAMED, see Article 29(2). Annex VI, Part C, Section 3.7 requires systems and procedure packs to bear their own UDI. For a procedure pack that appears quite straightforward. A separate label can be used on the procedure pack where these details can be entered, while the individual components keep their own labeling (there are exemptions for well known, single use devices or devices that are exempted to have UDI on their labeling, see Annex VI, Part C, Section 6.3.2).
Systems don�t have a single packaging, but their components can be placed on the market with reference to their specific use in this context. That means the system is placed on the market with multiple labels. Each label can refer to the same system UDI, with a separate UDI-PI for that component or set of components.
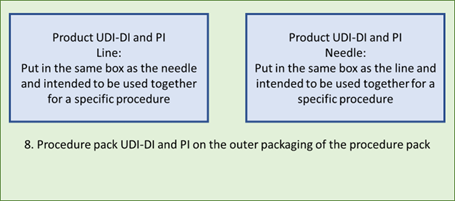
8. In the above examples this would work out as follows:8. Line and needle in a procedure pack: each has their own label with their individual UDI-DI and UDI-PI. On the procedure pack there is a UDI-DI and UDI-PI for this particular procedure pack.
9. Line and needle in a procedure pack, combined in a system with the pump: the intended purpose of the procedure pack should identify the pump and the UDI of the full system should also cover the combination of line and needle. This can be covered by a single system/procedure pack UDI-DI and UDI-PI on the procedure pack. The pump will have its own labeling and UDI, plus the same system UDI-DI and its own system UDI-PI.
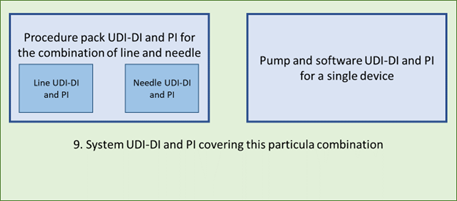
10. Line and needle in a procedure pack, combined with a system consisting of a pump with software: the system must identify all components involved (one component is a procedure pack) and cover that in one UDI-DI. Each component has the same system UDI-PI and their own system UDI-PI.
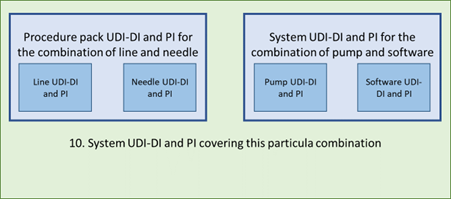
This example demonstrates that it can quickly become quite complex to identify all levels and forms of UDI. It is therefore recommended to develop a good understanding of how this works and make sure the UDI structure is checked thoroughly.
Legacy devices and Article 12 MDD or Article 22 MDR
Under the �old� Medical Devices Directive 93/42/EC (MDD) systems and procedure packs were regulated under Article 12. It would have made sense to apply Article 22 in full from the date of application of the MDR, but MDCG guidance 2021-25 on legacy devices allowed for a transitional arrangement. This results in the following:
- If the system and procedure pack is new and all devices are legacy devices, Article 22 MDR applies because Article 12 MDD is no longer valid.
- If the system and procedure pack has been identified under Article 12 MDD and at least one or the devices is certified under the MDR, Article 22 MDR applies. This includes MDR Class I devices.
- A new system or procedure pack with MDR certified devices is obviously fully ruled by Article 22 MDR.
System and Procedure Pack Producer: SPPP
Companies may not be aware they are placing devices on the market in combinations that qualify as systems or procedure packs, or their systems and procedure packs under MDD Article 12 should now be ruled by MDR Article 22. They should check this carefully. If this is the case, they must be aware that the SPPP is a specific role in EUDAMED and they must apply for a specific Single Registration Number (SRN).
Only with that SRN they can combine multiple UDI in EUDAMED. Legacy devices don�t have a UDI, but they have automatically been assigned a EUDAMED DI and EUDAMED ID to replace the Basic UDI-DI and UDI-DI respectively. Article 29(2) requires assigning a Basic UDI-DI and UDI-DI to an Article 22 system or procedure pack before placing on the market. Obviously, the role of SPPP should also be covered in their QMS. You can only arrange these EUDAMED DI�s and ID�s via EUDAMED. This implies that placing devices on the market under Article 22 requires registration in EUDAMED.
Article 22 is not covered by a notified body certificate, except in case of sterilization of a procedure pack (the sterilization process only). Therefore this is typically something that falls within the scope of the competent authorities and this will draw extra attention at the moment a system, procedure pack or a component is involved in a serious incident. In such situations it looks better if you are well prepared, so this is something to consider carefully and prepare well. As can be seen above, this can be quite complex and expert help may be recommended.
For assistance with structuring systems and procedure packs and other EUDAMED and UDI related questions, or even for just checking if Article 22 would have consequences for the current portfolio, just reach out. You can read more about grouping devices into a Basic UDI-DI here, and you can read about the strategic importance of the Basic UDI-DI here.



